







Collaborative Research Center (SFB) 677 - Function by Switching
Conferences
Third SFB Conference: Molecular Switches: Elementary Processes and Applications
August 27 - 30, 2017 at Plön Castle

Location
View Larger Map
Topics
Molecular Switches: Elementary Processes and Applications
- solid state and materials
- supramolecular chemistry
- STM / local manipulation
- molecular architectures
- ultrafast dynamics
- photochemistry and theory
- spin switching
Invited Speakers
- Jeremie Leonard (Strasbourg)
- Karl-Heinz Ernst (EMPA, Switzerland)
- Vincent Repain (Paris, Diderot)
- Christoph Lienau (Oldenburg)
- David Leigh (Manchester)
- Rafal Klajn (Weizmann)
- Giovanni Granucci (Pisa)
- Achim Schöll (Würzburg)
- Hermann Wegner (Gießen)
- Stefan Bräse (Karlsruhe)
- Christof Wöll (Karlsruhe)
- Nathalie Katsonis (Twente)
- Dr. Marc. H. Prosenc (Kaiserslautern)
Venue
- built 1633-1636 by Duke Joachim Ernst of Sonderburg-Plön
- Ducal residence since 1636
- Royal Danish summer residence since 1761
- Royal Prussian cadet academy since 1868
- Academy for the sons of Emperor Wilhelm II since 1896
Lectures
- invited lectures: 25 min + 5 min discussion
- internal lectures: 15 min + 5 min discussion
- conference language: English
Schedule
download schedule as PDF here:
show schedule
| Sunday, Aug 27 | ||
| 13:00 | arrival | |
| 16:00 | registration | |
| 18:00 |
welcome reception
award ceremony: KINSIS Award 2017 welcome party |
|
|
award lecture |
Dommaschk
|
Photoswitchable contrast agents for MRI |
|
show abstract insert abstract here |
||
| 19:30 | dinner | |
| Monday, Aug 28 | ||
| 07:00 | breakfast | |
|
09:00 |
Leonard
|
Vibrationally coherent optomechanical energy transduction in biomimetic molecular switches. |
|
show abstract Ultrafast C=C double bond photoisomerization converts light energy in mechanical energy at the molecular scale and may therefore be exploited in molecular devices for functional switching or rotary motion.[1] In the rhodopsin protein (Rh), the sensor for vision, the ultrafast photoisomerization of the protonated Schiff base of retinal (PSBR) triggers the protein activity. This photoreaction has outstanding speed and quantum yield and it appears to be vibrationally coherent, meaning that the energy of the absorbed photon is efficiently funneled into the isomerization coordinate on a time scale faster than energy dissipation to the environment. This unique property is the promise for an optimum photomechanical energy conversion. Following a biomimetic approach, the N-alkylated indanylidene– pyrrolinium (NAIP) molecular framework (see Figure 1A) was designed and synthesized such that its π-electron system would mimic that of PSBR in Rh. Consequently, its photoreaction dynamics in solution was shown to be very similar to that of Rh [2], including signatures of low-frequency vibrational coherence in the photoproduct ground state[3]. Here we apply a recently built experimental set-up utilizing sub-8fs UV-vis pulses to perform vibrational coherence spectroscopy [4] on the NAIP compounds. The objective is to reveal the signatures of the vibrational dynamics that drives the system through the conical intersection from the initial (Franck Condon) structure to the photoproduct. Importantly, the vibrational activity and photoreaction dynamics are critically influenced by the intramolecular steric hindrance imposed by a simple methyl substitution. Hence, we demonstrate that the appropriate chemical design may turn on or off this vibrationally coherent mechanism in the NAIP compounds. Computational quantum chemistry allows us to rationalize the underlying steric-electronic effect. We discuss the possible implications of this finding on the mechanism engineered by Rh to enhance the retinal isomerization yield, and on the future design of synthetic molecular switches targeting enhanced photoreaction yield.
References:
|
||
|
09:30 |
project B9
|
Functionalization and Investigation of Azo Functionalized TATA Platforms |
|
show abstract Upon irradiation with UV light (~365 nm) azobenzenes isomerize from their trans configurations to the corresponding cis isomers. The reaction back to the more stable trans isomer can either be performed by irradiation with visible light (~430 nm), or it proceeds spontaneously in the ground state. In homogeneous solution, the thermal cis to trans isomerization is slow at room temperature with half-lives of hours to days. The half-life of the parent azobenzene is 5 days at 25°C in benzene.[ R. J. W. Le Fevre, J. Northcott, J. Chem. Soc. 1953, 867] Similar values have been determined in other solvents, in the gas phase and in the melt.[ J.-A. Andersson, R. Petterson, L. Tegner, J. Photochem. 1982, 20, 17.] Very surprisingly, we observed half-lives of azobenzenes that are three orders of magnitudes faster (less than a minute) if the azobenzenes are connected via an ethyne spacer to a molecular platform which is absorbed on a Au(111) surface (see illustration). Since there is conjugation from the azobenzene through the triple bond into the system of the platform, we attributed this more than 1000fold rate acceleration to some unknown effect which is related to electronic coupling to the surface. To check this hypothesis, we gradually reduced conjugation by systematic variation of the spacer. Biphenyls are known to exhibit decreasing conjugation with increasing torsional angle between the two phenyl rings. This torsional angle in turn can be increased by introducing substituents in ortho position of the biphenyl unit. If conjugation is completely shut off (e.g. with two methyl groups) the cis-trans isomerization rate is almost the same in solution and on the surface confirming our preliminary hypothesis. 
[1] R. J. W. Le Fevre, J. Northcott, J. Chem. Soc. 1953, 867.
|
||
|
09:50 |
project A3
|
Spin Switching of Transition Metal Complexes |
|
show abstract Hair clip porphyrins combine the structural change and the ligand binding affinity of photoexcited nickel porphyrins into an intramolecular feedback.[1-3] This feedback should allow a new highly efficient photoswitching mechanism of molecular magnetic properties without utilizing photochromic groups. The rigid bridge between two meso-substituents prevents the intramolecular coordination of the linked ligand in the ruffled conformation of the low spin state. After photoexcitation the bridge is stretched by the flattening of the porphyrin and forces the incorporated ligand close to the nickel ion. Subsequently axial coordination occurs due to increased ligand affinity of the nickel ion in the high spin state and stabilizes the paramagnetic isomer. By excitation of the paramagnetic isomer the process should be reversible. Compared to azopyridines the intended switching mechanism requires minimal movement and does not rely on photochemical reactions like conventional photochromic switches.[4] Therefore switching should be feasible in solution as well as in solid state. Surface deposition is an established technique for porphyrins which extends the scope of the hair clip porphyrins. The transferability of the system between different environments opens up a variety of possible solution, surface and solid state analytics and applications. 
Scheme 1. Scheme of the intramolecular feedback in Hair Clip Porphyrins towards photoswitching.
Reference
|
||
|
10:10 |
Repain
|
Voltage and light induced spin switch of spin-crossover molecular monolayer on surfaces |
|
show abstract Spin-crossover molecules have the ability to change their spin state under external stimuli like temperature, pressure or light. So far, their switching properties, including cooperativity during the spin-crossover transition, had been measured mainly in their bulk state with techniques averaging over a large ensemble of molecules. Recently, few papers have shown the ability to preserve their spin-crossover properties when adsorbed on surfaces. Moreover, scanning tunneling microscopy allowed to induce and observe the switching of single or multiple molecules, either by pulses of electric field or by combination with light illumination. In this talk, I will recall the physics of spin-crossover molecules and review the recent works dealing with molecules on surfaces. |
||
| 10:40 | coffee break | |
|
11:30 |
project A7
|
Photo-Induced Dynamics of H-Transfer Switches |
|
show abstract Bistable intramolecular H-transfer systems exhibit a huge potential as photochromic molecular switches as they often feature electronic deactivation on an ultrafast timescale. Thus, undesired side reactions are virtually eliminated, thereby enabling record numbers of switching cycles. Within the scope of the Kiel CRC, special interest is drawn to such bistable intramolecular H-transfer switches in a joint effort of organic synthesis, theoretical calculations and spectroscopic experiments. Amongst others, we focus on a new design for photoreversible H-transfer switches, which has recently been proposed on the basis of theoretical quantum chemical calculations [A. L. Sobolewski, Phys. Chem. Chem. Phys. 2008, 10, 1243]. 
According to this scheme, photoexcitation initiates an ultrafast H-transfer in the excited state from a donor site to an intermediate acceptor group. Following a large-amplitude torsional motion driven by electronic deactivation through a conical intersection with the electronic ground state, the proton is finally dropped at its acceptor site. As a net result, the proton is transported along the molecular scaffold. Here, we report on recent results of femtosecond time-resolved spectroscopy, quantum chemical calculations and organic synthesis towards such bistable photochromic switches. Emphasis is put on the direct observation of the H-transfer pathway of N-(3-pyridinyl)-2-pyridine-carboxamide (NPPCA) on the sub-picosecond timescale by combined time-resolved fluorescence, electronic and vibrational absorption spectroscopy aided by theoretical calculations. In addition, first experimental and theoretical results will be reported for novel other H-transfer switches, including newly synthesized hydroxyquinoline-based ones. |
||
|
11:50 |
project B11
|
Photoswitching of Bacterial Adhesion |
|
show abstract Glycocalyx, the carbohydrate-rich layer lining the outermost part of the endothelium, is one of the most important and intriguing surfaces in biology. Molecular interactions between glycocalyx components and the carbohydrate-specific proteins (lectins) initiate multiple biological functions such as cell adhesion, immune response, and cell signaling. Adhesion of bacterial cells to their host cells also occurs through the specific binding of adhesive organelles of bacteria, known as fimbriae, with glycocalyx components present on the host cell surface. The aim of our project is to accomplish photoswitching of bacterial cell adhesion. With selective photoswitching of carbohydrate-specific bacterial adhesion, we aim to differentiate “good” (own) and “bad” (alien) cell adhesion with temporal and spatial resolution. So far, we have synthesized and studied mono- and disaccharide azobenzene glycosides and investigated the influence of sugar configuration and azobenzene substitution on their photochromic properties. 1,2 Furthermore, we have synthesized many multivalent azobenzene glycosides and have also shown how the versatile “click” chemistry can be used for this purpose.3 Our studies on the inhibitory potential of azobenzene mannosides in bacterial adhesion (E. coli with type 1 fimbriae, which possess an α-D-mannoside-specific lectin named FimH in its terminus) show that they are potent ligands of FimH. 1 Also, azobenzene mannosides with E or Z configuration bind approximately equally well to FimH, regardless of the configuration of the azobenzene N=N double bond. As a highlight of our project, we have successfully prepared photoswitchable SAMs (self-assembled monolayers) of azobenzene glycosides on gold surface.4 With these SAMs we have shown, for the first time, that threedimensional presentation of carbohydrates is critical in their interaction with bacteria.5 We found that when E azobenzene-conjugated mannoside derivatives on the gold surface were photoswitched to the Z form, the number of adherent bacteria (E. coli with type 1 fimbriae) decreased by a factor of five. We have also demonstrated that the carbohydrate-orientation based bacterial adhesion results are valid when azobenzene mannosides were immobilized on the surface of human cells too. 6 
Currently, we are trying to improve photoswitiching on surfaces and also to understand the structure-function relationships that underlie the observed biological effects. To that end, we plan to prepare “self-diluting” systems with molecules that occupy space during the assembly process, which can, in a consecutive step, be released by cleavage “on SAM”. Also, we will synthesise azobenzene glycoclusters, which will permit to differentiate the effect of “scaffolded multivalency” from “surface-based” multivalency. In another approach towards better photoswitchable glycoarrays we plan to use a concerted “enforced” E/Z isomerization of glyco- SAMs with molecules possessing a rigid backbone.1 Finally, in order to explore the effect of carbohydrate orientation in bacterial adhesion in a relatively complex glyco-SAMs, we will prepare mixed SAMs with two different types of azobenzene glycosides. The two ligands will be chosen in such a way that they undergo photoisomerization under different wavelengths and adhere to E.coli strains with different carbohydrate specificity. These orthogonal glycoSAMs will be a better mimic of glycocalyx and will take us a step closer towards understanding the functioning of glycocalyx. On another front, in order to achieve photoswitiching of bacterial adhesion in solution, the adhesive protein FimH will be modified site-specifically to make ligand binding switchable by reversible blocking of the carbohydrate binding site of the lectin. Modification of the protein FimH shall result in installation of a photoswitchable azobenzene derivative to act as a “gate keeper” molecule for reversible blocking of the entrance of the lectin’s carbohydrate binding site. This will allow photoswitching of bacterial adhesion in solution, independently of a modified surface. Our studies on photoswitching of bacterial adhesion both on glyco-SAMs and in solution will provide a better understanding of the bacterial adhesion mechanism along with shedding more light into the functioning of glycocalyx. Acknowledgement: Our collaborations with the Tuczek (Kiel) and the Terfort (Frankfurt) groups are gratefully acknowledged.
References
|
||
|
12:10 |
Wöll
|
Liquid Phase Epitaxy of Molecular Frameworks on Solid Substrates: A New Class of Designer Solids? |
|
show abstract With regard to the development of approaches to realize “Designer Solids” by programmed assembly of building units taken form libraries, recently metal-organic frameworks (MOFs) have attracted a huge interest. Here, we will focus on MOF-based electrochemical [1,2], photoelectrochemical [3] and photovoltaic devices [4,5]. Internal interfaces in MOF heterostructures are also of interest with regard to photon-upconversion [6] and can be used for the crosslinking of sandwiched, reactive monomers [7]. Since the fabrication of reliable and reproducible contacts to MOF-materials represent a major challenge, we have developed a layer-by-layer (lbl) deposition method to produce well-defined, highly oriented and monolithic MOF thin films (SURMOFs) on a number of different substrates. 
At present, the whole field is reaching a rather mature state, with a number of functioning SURMOF-based devices, and many other proposed [10]. In the talk, we will present a few examples to demonstrate the state of the art and the main challenges. Furthermore, we will discuss the potential of these „Designer Solids“ to construct host matrices for studying molecular interactions, and in particular the switching of photoactive compounds.
References:
|
||
| 12:40 | lunch | |
| 14:00 | poster session | |
|
16:00 |
Leigh
|
Making the Tiniest Machines |
|
show abstract Over the past two decades some of the first examples of synthetic molecular level machines and motors—all be they primitive by biological standards—have been developed. Perhaps the best way to appreciate the technological potential of controlled molecular-level motion is to recognise that nanomotors and molecular-level machines lie at the heart of every significant biological process. Over billions of years of evolution Nature has not repeatedly chosen this solution for achieving complex task performance without good reason. When we learn how to build artificial structures that can control and exploit molecular level motion, and interface their effects directly with other molecular-level substructures and the outside world, it will potentially impact on every aspect of functional molecule and materials design. 
Selected papers:
|
||
|
16:30 |
project C1
|
Light-Induced Conductance Switching in Photomechanically Active Carbon Nanotube-Polymer Composites |
|
show abstract Novel optically responsive devices with hosts of potential applications have been demonstrated by coupling carbon nanomaterials with photochromic molecules. For lightinduced conductance switching, in particular, azobenzene containing carbon nanotubepolymer nanocomposites proved to be very attractive, providing stable and no-degradable changes in conductivity over time at standard laboratory conditions. In these composites, the photoswitching mechanisms are based on light-induced changes in electronic properties and related to the Pool-Frenkel conduction mechanism. However, no link between conductivity switching and the molecular motion of azobenzene chromophores could be found, due to application of high elastic modulus polymer matrices. Here we report on single wall carbon nanotube-polymer nanocomposites with a soft polycaprolactone polymer host. This system clearly shows the transfer of light induced nano-sized molecular motion to macroscopic thickness changes of the composite matrix. We demonstrate that these photomechanic effects can indeed dominate the conductivity switching behavior over the electronic effects and lead to a reversion of the switching direction near the percolation threshold. |
||
|
16:50 |
project B13
|
NEXAFS study of novel robust spin crossover films on metallic surfaces |
|
show abstract The spin-state manipulation of Fe(II) coordination complexes by external stimuli is associated with a change in the electronic, magnetic, and structural properties. In particular, the temperature- and lightinduced switching mechanisms may be utilized in future spintronic devices. Both of these switching effects were observed even in single monolayers adsorbed on the weakly interacting substrate HOPG[1]. Nevertheless, the dissociation of spin-crossover (SCO) molecules in direct contact to a metal surface is still impeding measurements in device-like environments. A novel approach to overcome this limitation is the electronically stabilized SCO system Fe(PyPyr(CF3)2)2(phen), a recently synthesized derivative of the well-studied complex Fe(H2B(pz)2)2(phen)[2,3]. Here, we present near edge X-ray absorption fine structure (NEXAFS) spectroscopy results of ultrathin films of Fe(PyPyr(CF3)2)2(phen) adsorbed on Au(111) and TiTe2, for different temperatures and laser light irradiation conditions. Our results confirm a complete and reversible light-induced spin-crossover in thin films of this derivative, prepared under ultra-high vacuum conditions. 
Fig. 1: Fe-L-NEXAFS spectra of Fe(PyPyr(CF3)2)2(phen) on TiTe2 at various temperatures during 532 nm laser light irradiation.
[1] M. Bernien, H. Naggert, L. M. Arruda, L. Kipgen, F. Nickel, J. Miguel, C. F. Hermanns, A. Krüger, D. Krüger, E. Schierle, E. Weschke, F. Tuczek, and W. Kuch, ACS Nano 9, 8960 (2015).
|
||
|
17:10 |
Wegner
|
Azobenzene switches – from physical organic chemistry to energy storage |
|
show abstract The synthesis and control of structure on the molecular level allows designing properties to address fundamental questions in chemistry and to develop new materials. Azobenzene switches have been applied extensively to control functionality on the molecular level. However, the understanding of the switching processes is still an active field of research. The incorporation of azo units within macrocycles, for instance, allows to probing their inherent properties. Additionally, azobenzene switches have been applied to investigate one of the most basic interactions in chemistry - the attractive London-dispersion as part of the van-der-Waals forces. Finally, the potential for materials applications is discussed. 
Angew. Chem. Int. Ed., 2015, 54, 13436-13439. Org. Biomol. Chem. 2014, 12, 3371. |
||
| 19:30 | dinner | |
| Tuesday, Aug 29 | ||
| 07:00 | breakfast | |
|
09:00 |
Ernst
|
Helical molecules at surfaces: selective chemistry and molecular machines |
|
show abstract Molecular recognition among chiral molecules on surfaces is of paramount importance in biomineralization, enantioselective heterogeneous catalysis, and for the separation of chiral molecules into their two mirror-image isomers (enantiomers) via crystallization or chromatography. Understanding the principles of molecular recognition in general, however, is a difficult task and calls for investigation of appropriate model systems. One popular approach is thereby studying intermolecular interactions on well-defined solid surfaces, which allows in particular the use of scanning tunneling microscopy (STM). Examples of chiral amplification via the so-called ‘sergeant-and-soldiers’ effect as well as manipulation of chiral adsorbates via inelastic electron tunneling will be presented. Moreover, we report spin-dependent filtering of electrons by monolayers of these helical molecules. Finally the first successful electrical current-driven, unidirectional motion of a synthetic molecule on a surface will be presented. |
||
|
09:30 |
Lienau
|
Ultrafast multidimensional spectroscopy of charge-transfer processes in light-harvesting systems |
|
show abstract The efficient conversion of (sun-)light into electrical or chemical energy is one of the most fundamental processes in biology and at the same time a highly-relevant challenge in current energy research. Generally, it is well understood that the underlying microscopic conversion processes happen on an exceedingly short femtosecond time scale and typically involve the transfer of charge from an optically bright donor moiety to an optically dark acceptor system. What is less clear is whether vibronic quantum coherence is helpful or maybe even necessary for those processes to occur and to what extent vibronic quantum coherence modifies the transport of charges through these systems. In my talk, I want to give a few examples how ultrafast, two- and three-dimensional optical spectroscopy – combined with advanced theoretical modelling - can shed new light on those questions how it can provide quite detailed insight into quantum-coherent photoinduced charge separation processes [1-3]. Specifically, I want to discuss the possible role of conical intersections on these processes.
[1] C. A. Rozzi, S. M. Falke, N. Spallanzani, A. Rubio, E. Molinari, D. Brida, M. Mauri, G. Cerullo, H. Schramm, J. Christoffers, and C. Lienau, Quantum coherence controls the charge separation in a prototypical artificial light harvesting system, Nature Communications 4, 1602 (2013).
|
||
|
10:00 |
project C12
|
A Photoswitchable Kinase Inhibitor: Photoinduced E/Z Isomerization of Axitinib |
|
show abstract Protein kinases are enzymes that mediate signal transduction in intracellular signal pathways and regulate cell growth and differentiation. Overactivated kinases, however, can lead to uncontrolled cell proliferation and play a crucial role in tumor progression and inflammatory diseases. Therefore, kinases are drug targets and the development of small molecule kinase inhibitors has become a major field in pharmaceutical research. [1] Our goal is to develop photoswitchable kinase inhibitors that can be spatially and temporally controlled by light. These compounds will be useful not only for innovative therapeutic approaches but also as novel pharmacological tools, e.g. for resolving dynamic aspects of kinase signal transduction. 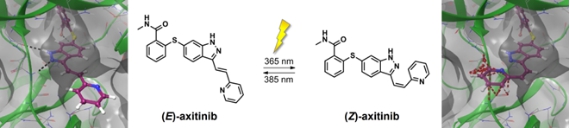
Scheme 1. (Left): (E)-axitinib in the ATP binding pocket of VEGFR2 (pdb code 4AG8). [2]. (Right): Superposition with (Z)-axitinib (steric clashes with the protein are indicated as red dotted lines). In the present study, we focused on the protein kinase inhibitor axitinib which is approved for advanced metastatic renal cell carcinoma (RCC) since 2012. Interestingly, axitinib undergoes an E-Z isomerization upon irradiation with UV light (Scheme 1). [3] Hence, our goal was to explore if its inhibitory effect can be turned “on” and “off” triggered by light. Indeed, we could demonstrate that (Z)-axitinib is up to 43 times less active in an in vitro VEGFR kinase assays and 19 times less active on human umbilical vein endothelial cells (HUVEC) compared to (E)-axitinib. By irradiating (Z)-axitinib with UV light (385 nm) it is possible to switch it completely to the (E)-isomer and to restore the biological activity of (E)-axitinib. However, vice versa it is not possible to switch the biological activity of (E)-axitinib “off” in aqueous solution.
Reference
|
||
|
10:20 |
project A1
|
Ultrafast Photo-induced Dynamics of the Magnetically Bistable Azopyridine/Ni-Porphyrin Record Player Spin Switch |
|
show abstract The azopyridine-functionalized Ni-porphyrin 1 [Venkataramani et al., Science 2011, 331, 445; Fig. 1] shows magnetic bistability in homogeneous solution at room tempe- rature. On irradiation at λ ~ 500 nm, the four-coordinate low-spin (LS) Ni switches to its high-spin (HS) state, which is stabilized by axial coordination of the azopyridine ligand that undergoes trans-to-cis (E-to-Z) isomerization. At λ ~ 435 nm, this process is reversed and the LS state is efficiently recovered. 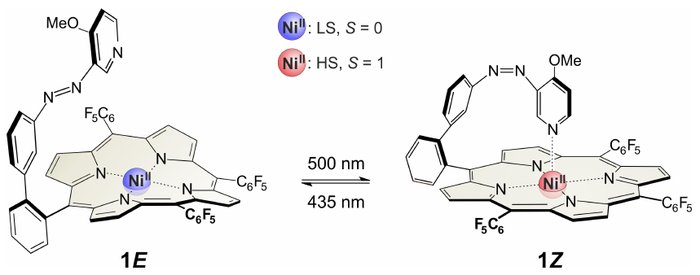
We have investigated the ultrafast dynamics of the E-LS and Z-HS states of 1 using femtosecond transient electronic absorption spectroscopy after photoexcitation in the Q band or in the B band of the porphyrin. The four-coordinate E-LS species 1E was found to exhibit virtually identical dynamics as the plain Ni-porphyrin. Transient switching to the LS state happens in <0.25 ps, but E-to-Z isomerization of the azopyridine arm and stabilization of the HS state in the form of 1Z is very inefficient, making this a very minor channel. The five-coordinate Z-HS compound 1Z, on the other hand, also shows virtually immediate spin switching in <0.25 ps, subsequent vibrational cooling in ~12 ps, followed by a persistent weak residual LS product absorption lasting beyond Δt = 1 ns. The measured quantum yields are, however, very low (ΦHS→LS = 0.06 0.01, Φ1Z→1E = 0.02 - 0.03; vs. Φ1E→1Z < 0.01). The photo- induced dynamics are dominated by coupling of the porphyrin ππ* and Ni(d-d)/Ni(d2) states, not by initial photo-isomerization of the azopyridine. The very minor Z-E isomerization of the ligand might instead take place via the azopyridine triplet state. |
||
| 10:40 | coffee break | |
|
11:30 |
Katsonis
|
Photo-mechanizing soft matter with molecular switches |
|
show abstract The sophistication reached by organic chemistry has enabled the design and synthesis of a wide range of dynamic molecules that display controlled shape changes with an ever-increasing refinement. However, amplifying these molecular-scale dynamics to support shape-transformation, motility, and eventually a broad range of macroscopic functions remains a key challenge. I will discuss how we draw inspiration from the operational principles of living materials in order to address this challenge, with a special focus on coupling the operation of molecular photo-switches to liquid crystal networks. Besides shape transformation and autonomous motion, we seek to pre-program adaptive mechanical properties at the macroscopic level. |
||
|
12:00 |
Schöll
|
Orbital Imaging by Angle Resolved Photoelectron Spectroscopy: From Basic Principles to High Resolution Experiments and Limitations |
|
show abstract In recent years angle-resolved photoelectron spectroscopy (ARPES) has emerged as a particularly powerful tool for the imaging of orbitals [1,2]. In this talk I will briefly introduce this technique and discuss its prospects and main limitations. The imaging of orbitals in momentum space by ARPES has been applied in several instances, e.g. to show hybridization at interfaces [3], the resulting band dispersion [4] and many particle effects [5,6]. By retrieval of the phase information, which can be done by mathematical algorithms [7] or by experiments with circular light polarization [8] orbitals can also be reconstructed in real space and in three dimensions [9]. In particular by using state-of-the-art momentum microscopes with high energy resolution in combination with synchrotron radiation orbital imaging by ARPES can provide unprecedented insight into the properties of molecular materials. For example, photoelectron momentum microscopy with high energy resolution allows imaging of molecular orbitals with resolution of vibronic modes. I will demonstrate that the intensity patterns of photoelectrons derived for the vibronic sidebands of molecular states show characteristic changes due to the distortion of the molecular frame in the vibronically excited state (see Figure 1). By a comparison to the simulated patterns derived from calculations, an assignment of the specific vibronic mode that preferentially couples to the electronic excitation is possible, which in the example of the highest occupied molecular orbital (HOMO) of coronene is a b2u inplane mode with an energy of 0.2 eV [10]. 
Figure 1: a) Energy distribution curve recorded for a monolayer of coronene on Au(111). The spectrum was fitted by a vibrational progression consisting of three equally spaced Voigt peaks. Photoelectron momentum maps of the HOMO of coronene/Au(111) recorded at the energy of the (0-0) peak (b) and of the vibronic side bands ((c) and (d)). e) Illustration of the Ag-mode responsible for the sidebands. (from [10]). Orbital imaging by photoelectron momentum mapping with vibronic resolution thus provides unique information for the analysis of the coupling between electronic and vibronic excitation. Limitations of the technique are mainly imposed by the plane wave final state approximations. As I will demonstrate, this is not sufficient for certain experimental conditions, three dimensional objects, or if scattering cannot be neglected. In such cases more elaborated calculations such as time-dependent DFT [11] or the Independent Atomic Center approximation can provide a more exact description of the ARPES data.
References
|
||
|
12:00 |
project A8
|
Function by Switching: Molecular Assemblers, Molecular Machines Performing Synthesis |
|
show abstract In chemical synthesis usually the reactants are dissolved in an organic solvent, the reactive molecules undergo stochastic collisions and form a bond if kinetic energy and relative orientation are favourable. However, the majority of biologically active molecules in nature are synthesized in ATP driven, molecular machine-type enzyme complexes such as non-ribosomal peptide synthetases (NRPS) or polyketide synthases (PKS). They operate like an assembly line by guiding reactions under positioning control driven by ATP. Notwithstanding the fact that there are a number of advantages to this assembler-like synthesis (less side reactions, easy stereo control, no protecting groups, preselection of reactants, driving unfavourable reactions…), there is (according to the best of our knowledge) no artificial system published so far. We are aiming at the design, synthesis and investigation of the first model system of a molecular assembler. In our preliminary work we designed and synthesized a light-switchable ditopic receptor which is able to drive the condensation of 4 molecules of vanadate to a cyclic tetravanadate. The reaction which is endergonic and therefore not spontaneous in the absence of the ligand is driven by the large and selective binding energy of the product tetravanadate inside the receptor. Photochemical isomerization (365 nm) of the ligand releases the product. Upon irradiation with 430 nm the original, “empty” state is restored and the cycle starts again. 
|
||
| 12:30 | lunch | |
|
14:00 |
Prosenc
|
Covalently Linked Magnetic Complexes on Surfaces: Spin Delivery, Logic Devices and Photo-Switches |
|
show abstract Molecular spintronic devices offer great potential for future energy-efficient information technology as they combine ultimately small size, high-speed operation, and low-power consumption. Recently, atom by-atom assembly together with spin-sensitive imaging and characterization at the atomic level have led to a first prototype of an all-spin atomic-scale logic device [1]. One major disadvantages of such atom based spintronic device is its low thermal stability of the assembly. By employing tailor-made paramagnetic molecular building blocks, combined with deposition under ultra-high vacuum conditions as well as surface chemistry[2], a corresponding prototype of an all-spin based molecular device can be realized. 
Such molecular device concepts offer the advantage of inherent parallel fabrication based on molecular self-assembly as well as considerably higher operation temperatures due to enhanced energy scales of covalent through-bond linkage of basic molecular units compared to substrate-mediated coupling schemes employing indirect exchange coupling between individual adsorbed atoms on surfaces. Further advantages are, that the molecular end electronic properties of the molecules remain which extend the scope of molecular based devices.
[1] A. A. Khajetoorians, J. Wiebe, B. Chilian, R. Wiesendanger Science 2011, 332, 1062-1064.
|
||
|
14:30 |
project C14
|
Polymer Composite Inspired by the Human Skin: Molecular Switches in Complex Environments - Progress Report on Mechanophoric Composites |
|
show abstract Molecular switches such as azobenzene, spiropyran and spirooxazine have been incorporated into several complex environments for example azobenzene in liquid crystalline polymer for a photo-mechanical effect and spiropyran in polymer chains as a multi-stimuli component. Here presented is a research on direct and indirect incorporation of the molecular switches in various polymers. Spiropyran and spirooxazine were used as fillers; by simply dispersing it into the chemically stable and solvent free polythiourethane (PTU) multi-stimuli composites were produced. A very efficient UV sensor existing in the nature is human skin. Inspired by the human skin a polymer composite based on a photochromic spirooxazine is part of the presented work. Molecular switches used here are the molecules that undergo a well known spiro-mero reversible isomerisation under UV light indicated by a color change. The isomerisation is reversed either by visible light or by thermal relaxation. When the photochromic polymer composite was exposed to the direct sunlight it turned purple. The PTU/spirooxazine composite was then partially covered with a sunscreen spray (SPF 50+) and was placed under sunlight. As the sunscreen fade the exposed colorless polymer composite turns purple as shown in the figure 1. An objective of the project C-14 was to covalently integrated spiropyran into the polymeric chains of polymethylacrylate (PMA), polymethylmethacrylat (PMMA) which was later coated around the glass fibers and T-ZnO to enhance the mechanical stability of the matrix and to obtain a self-reporting mechanophoric composite. Mutli-stimuli fibers were produced by spinning the polymer PMA-SP-PMA with the help of a self-made centrifugal spinner. 
Figure 1: Images of a photochromic polymer composite undergoing reversible isomerization when exposed to sunlight. (a) The PTU/photochrome composite completely covered with a sunscreen spray under the sunlight. (b) The composite partially covered with a sunscreen spray under sunlight. (c) The composite without sun protection under the sunlight. |
||
|
14:50 |
Bräse
|
Design and synthesis of novel molecular switches - from small molecules to materials |
|
show abstract In this talk, various new materials using supramolecular and/or covalent linking from various symmetrical organic building blocks are presented. Photoswitching using azobenzenes and other photoswitches will be discussed in Terms of design, Synthesis, efficiancy and applications. |
||
| 15:30 | departure | |
| 16:00 | boat excursion | |
| 19:30 | conference dinner | |
| Wednesday, Aug 30 | ||
| 07:00 | breakfast | |
|
09:00 |
Klajn
|
Reversible photochromism in the cavities of flexible coordination cages |
|
show abstract Placing various molecules in confined spaces can profoundly alter their physicochemical properties. Examples in nature are abundant and range from stabilizing otherwise unstable species (such as sulfenic acids1 ) to orienting molecules in ways that can greatly accelerate chemical reactions between them (such as the >107 rate acceleration of CO2 hydrolysis by carbonic anhydrase2 ). Over the past decade, chemists have investigated the behavior of different chemical species within synthetic confined environments. Notable examples include unusual regioselectivity and efficient catalysis of the Diels-Alder reaction within the cavities of octahedral palladium-based self-assembled cages,3 rendering white phosphorus stable to air,4 and promoting cationic reaction cascades leading to efficient terpene cyclizations.5 Inspired by the fascinating process of retinal photoisomerization within the cavity of rhodopsin,6 we are broadly interested in the behavior of photoswitchable molecules under confinement.7-10 Here, I will present our latest studies on the encapsulation of photoswitchable molecules within flexible self-assembled cages based on imidazole-palladium coordination. We found that the uptake of spiropyran by our cages is accompanied by its isomerization to its otherwise unstable, ring-open (merocyanine) form. The reaction proceeds quantitatively even for spiropyrans lacking the nitro group typically used to stabilize the open form. The encapsulation renders spiropyrans responsive to blue light, and the light-induced ring closing/spontaneous (dark) ring opening sequence can be repeated multiple times. This unexpected behavior of spiropyran within the cavities of these cages enabled us to fabricate gels, in which images could be created with light. Moreover, we found that a spontaneous conversion of the merocyanine form (blue) to the protonated merocyanine form (faint yellow) occurred spontaneously upon the dehydration of the system. This finding inspired us to prepare paper in which writing can be performed with water as the ink. We have also demonstrated an efficient uptake of various azobenzenes using the same flexible cages. Interestingly, azobenzene molecules were encapsulated as dimers, as confirmed by X-ray crystallography. This observation paves the way to studying the photoswitching behavior of discrete oligomers of photoswitchable molecules (here, dimers) within confined spaces.
References
|
||
|
09:30 |
project B12
|
Light-Triggered Control of Plasmonic Refraction and Group Delay by Photochromic Molecular Switches |
|
show abstract An interface supporting plasmonic switching is prepared from a gold substrate coated with a polystyrene film (PS) doped with spirophenanthrooxazine (SPO). A reversible light-induced change in the surface plasmon polariton (SPP) dispersion curve of the interface is experimentally demonstrated evidencing reversible switching of surface plasmon polariton group and phase velocity [1]. The switching capabilities of the interface are furthermore successfully applied to achieve focus control of a plasmonic lens. The results imply the realization of non-volatile and reversible plasmonic switching units providing complex functionalities based on surface plasmon refraction and group delay. 
Figure 1: 2P-PEEM images of the SPP intensity pattern next to the elliptic coupling edge of a model plasmonic lens as mapped with 740 nm laser light before (unswitched) and after (switched) illumination of the PS/SPO/gold sample with UV light (λ = 365 nm); the curved periodic pattern indicates the focusing of the SPP field. A clear shift of the focal point upon switching is observable. The dashed line indicates the position of the coupling edge.
References
|
||
|
09:50 |
project A6
|
Absolute Temperature Magnetic Resonance Imaging using Photo-Switchable Contrast Agents. |
|
show abstract Temperature Magnetic Resonance Imaging (MRI) is used for various applications, e.g. tissue ablation using high intensity focussed ultrasound [1] or radio frequency applicators for hyperthermia [2]. For these applications the proton resonant frequency shift (PRF) method [3] is used. Besides PRF techniques based on T1-relaxivity [4] or diffusion [5] exist. All MR intrinsic methods require reference images and can only be used for relative measurements. Temperature is a clinical marker for increased tissue metabolism and can be used to detect tumours or tissue inflammation [6]. Some temperature MRI contrast agents (CA) exist but they lack of image resolution [7] or use time-consuming MRI spectroscopy [8]. To increase image resolution and shorten measurement time a photo-switchable record player CA prototype was developed. The CA is based on a Ni(II)-porphyrin which is linked covalently with a photo-switchable azoimidazole at one of the meso positions. The axial azoimidazole is the tone arm of the so called record player molecule and binds in its cis form to the Ni(II)-ion. The CA can be switched on (to cis form) by light with a wavelength of 365 nm. In the cis configuration the complex is paramagnetic (high-spin, S = 1) and influences the relaxation times of its solvent (here: MeOH). The isomerisation process back to the trans configuration is only temperature dependant. Calibration measurements with the photo-switchable CA at different temperatures (-7°C to 7°C) were performed in a small animal MR system (ClinScan 7T, Bruker, Ettlingen, Germany) using a cooling jacket with different inversion recovery sequences. For reference a fiber-optical temperature probe (OpSENS, Ville de Québec, Canada) was placed inside the cooling jacket. During imaging the inversion time was set to the off-state of the solution suppressing signal of the solvent. Different CA concentrations were used to determine possible concentration dependencies. To create a temperature gradient and to measure absolute temperature with during standard imaging a custom-built cooling jacket was used. For this purpose the CA and a gel forming agent was dissolved in MeOH to suppress convective flow of heat. A temperature gradient of -5°C to 5°C was used. For imaging a 2D-IR-HASTE sequence with following imaging parameters was used: TR = 10 s, TE = 56 ms, TI = 2.3 s, FoV = 32 × 32 mm², Mtx = 256 × 256 px, Res =125 × 125 µm², SL = 1.1 mm, Repetitions: 360. To increase SNR a temperature map with 144 × 144 px was calculated (222 × 222 µm). Figure 1 shows the mono exponential signal decay at different temperatures (a) and different concentrations (b). The switching of the CA back to its ground state is only temperature dependent. 
Figure 1: a) MR signal decay at different tem-peratures. b) MR signal decay at different concentrations. Figure 2 shows the temperature map of rate constants of the pixel-wise fitted mono-exponential functions. The absolute temperature image clearly shows the gradient from both cold (-5°C) and warm (5°C) compartments of the phantom. With this developed CA it would be possible to perform temperature MRI with standard MR IR sequences during a standard clinical protocol. 
Figure 2: Temperature map calculated from 360 IR HASTE images. Due to low SNR (<10) the resolution of the temperature map was decreased by a factor of 1.8. For future developments water solubility and increasing the temperature range are planned to perform in vivo experiments.
[1] Krafft AJ et al. Med Phys 2010, 1: 2380-2393
|
||
|
10:10 |
project C13
|
Structure and Kinetics of Switching in Phospholipid Membranes |
|
show abstract Having integrated Amphiphilic photoswitchable molocules into phospholipid membranes, we investigate their biologically relevant properties. By studying the interactions in such a model system consisting of switchable biomimetic molecules in lipid membranes here, we study the structure and kinetics of membrane response to the switching process. These in situ experiments not only contribute to the fundamental understanding of membrane dynamics but also will contribute to potential applications for molecular switches such as drug delivery. In order to investigate these properties we study model systems in which amphiphilic photoswitchable molecules are integrated into Langmuir films of phospholipids. We have modified glycolipids to contain an azobenzene photoswitch between the chain and the head group and successfully embedded those in a monolayer of Dipalmitoylphosphatidylcholine (DPPC). This allows us to reversibly change the azobenzene-glycolipid orientation between trans- and cis-conformation by illumination with UV and blue light. We have followed the structural changes in this model membrane and the switching kinetics of the system with Langmuir isotherms and in situ X-ray methods, X-ray reflectivity and grazing incidence diffraction. The membrane structure responds on a time scale of seconds upon UV illumination. Switching from trans to cis conformation results in changes in molecule tilt and spacing and also membrane thickness. A critical point in surface pressure at 18.4 mN/m has been discovered above which the membrane expands rather than compresses upon switching from trans- to cis- conformation. |
||
| 10:30 | coffee break | |
|
11:00 |
projects B6, B7
|
Switching of single molecules on metallic surfaces |
|
show abstract Molecular electronics aims at utilizing functional molecules as building blocks of electronic components. In that respect, molecules adsorbed on a surface presenting a switching functionality are particularly interesting. Such molecules are however generally fragile and the deposition of such molecules onto surfaces may result in their decomposition [1]. Molecules possessing different properties were deposited on metallic surfaces using two different techniques: sublimation for the most robust molecules and electrospray deposition [2] for the more fragile molecules. The adsorption, along with the electronic properties of the deposited molecules, were investigated using low-temperature scanning tunneling microscopy and spectroscopy. The switching of the molecules was triggered by different means, e.g. electrons, mechanical forces and adsorption of ligands, resulting in changes of molecules’ properties. Indeed, the switching of molecules on surfaces can strongly affect their conformation [3], their adsorption properties [4] and/or their electronic and magnetic properties [5-7]. A strong emphasis will be given on the investigation of spin-crossover molecules, for which the switch is associated to concurrent conformational, electronic and magnetic changes [8]. Interestingly, the functionality of the molecules may initially not be present in the molecule but acquired upon adsorption on the surface. Finally, in perspective to integration of molecules into electronic devices, the conductance through single molecules is thoroughly investigated. In particular, it will be shown that the contact geometry strongly influences the junction conductance [9].
References:
|
||
|
11:20 |
project B10
|
Anisotropic magnetoresistance at the single molecule level |
|
show abstract The anisotropic magnetoresistance (AMR) is a phenomenon that is well known for bulk ferromagnets, however, it constitutes an effect of only a few percent. Here, we study the AMR for single molecules in the contact and in the tunneling regime based on density functional theory. In junctions containing single molecules sandwiched between ferromagnetic leads we show that the AMR can be enhanced by orders of magnitude with respect to bulk ferromagnets. We study ballistic transport in metal-benzene complexes contacted by 3d transition-metal wires. We show that a gigantic AMR can arise from spin-orbit coupling (SOC) effects in the leads, drastically enhanced by orbital-symmetry filtering properties of the molecules [1]. This molecular AMR (MAMR) can explain recent break junction experiments [2]. Scanning tunneling spectroscopy allows to measure the AMR in the tunneling regime for Pb dimers adsorbed in different orientations on the Fe double layer on W(110) [3]. Only for straight Pb dimers along the [001] surface direction a large tunneling AMR (TAMR) of about 20% is observed while no effect occurs for tilted Pb dimers. We explain this effect based on symmetry arguments and SOC induced orbital mixing as confirmed by DFT calculations [3].
[1] F. Otte, S. Heinze, and Y. Mokrousov, Phys. Rev. B 92, 220411 (R) (2015).
|
||
|
11:40 |
McConnell
|
Transformation of an Iron(II) Cage from High- to Low-Spin Switches Guest Release in a Two Cage System |
|
show abstract Metal-organic cages function as molecular containers as there is the potential to encapsulate guests with high affinity and selectivity within their well-defined cavities.1 Subcomponent self-assembly exploits the reversible formation of covalent and coordinative bonds to prepare metal-organic cages from relatively simple subcomponents. A wide variety of architectures from tetrahedra to icosahedra can be self-assembled through the formation of imine bonds around metal centres. There is growing interest in stimuli-responsive cages, whose properties (e.g. guest uptake/release) are altered in response to an external signal, in order to construct molecular networks approaching the complexity and functionality exhibited by signalling pathways in biological systems.2 We report spin state switching of a cage as a new strategy for signal transduction; the transformation of a high-spin FeII4L4 cage to its low-spin analogue is exploited to switch guest release in a two cage system.3
References
|
||
|
12:00 |
Granucci
|
Simulating the photoisomerization of self assembled monolayers of azobiphenyls |
|
show abstract We performed a computational investigation of the photodynamics of self assembled monolayers (SAM) of biphenyl azocompounds bound through a terminal S atom to a Gold surface. The trans and cis SAMs can be interconverted by irradiating with appropriate wavelengths and exhibit quite different structural, optical and electric properties [1, 2, 3]. The photoisomerization shows a cooperative behavior: in a partly isomerized SAM large domains of pure cis or trans isomers are found [1]. The nonadiabatic dynamics was simulated by the surface hopping method with quantum decoherence corrections. The electronic energies and wavefunctions were computed on the fly with the semiempirical FOMO-CI method in its QM/MM version [4]. Our results show that cis → trans photoisomerization occurs in all the environments. Conversely, trans → cis photoisomerization is suppressed in a pure trans SAM, while it occurs in single trans molecules embedded in a cis SAM. These effect of close packing can explain the previously observed cooperative behavior [1]. Other simulations highlights the role of defects in starting the trans → cis conversion: for example, in a trans SAM with a single cis molecule, the first neighbors of the cis molecule are photoreactive[5]. Moreover, concerning the suppression of photoisomerization, the steric confinement effect appears to be more important than exciton transfer [6].
[1] G. Pace et al, PNAS 104, 9937 (2007).
|
||
| 12:30 | closing remarks | |
| 12:45 | lunch | |
| 14:00 | departure | |